
Cada 23 de junio desde hace casi una década se conmemora el Día Internacional del Síndrome de Dravet, también conocido como Epilepsia Mioclónica Severa de la Infancia (SMEI), el cual fue descrito en 1978 por la psiquiatra y epileptóloga Charlotte Dravet.
Desde 1989 la Liga Internacional contra la Epilepsia (ILAE) lo incluye dentro del apartado de “Epilepsias y síndromes indeterminados respecto a la localización con crisis generalizadas y focales”.
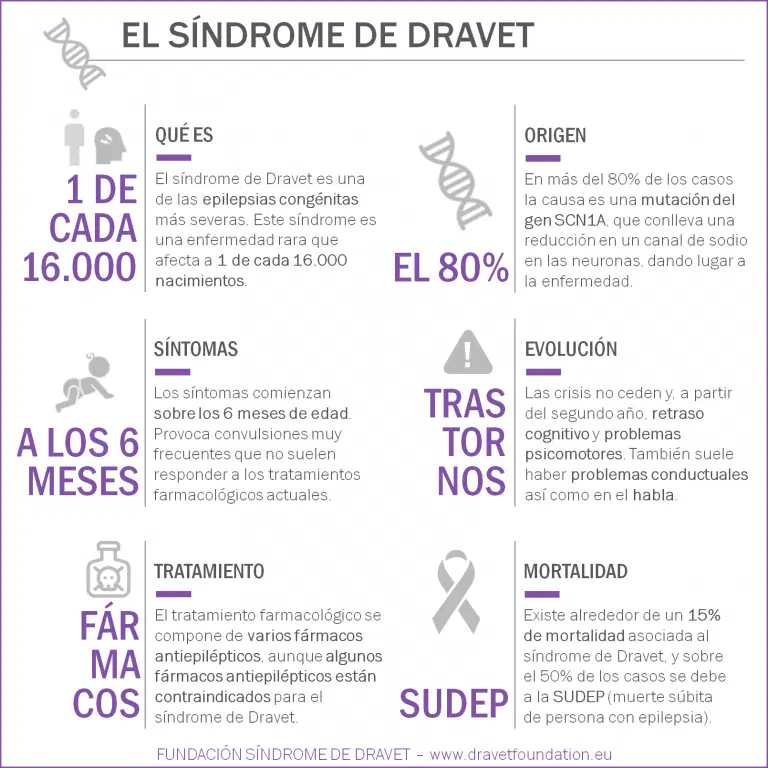
Se trata una encefalopatía epiléptica del desarrollo de origen genético y se encuadra dentro de la familia patológica de las canalopatías, ya que aproximadamente el 80% de los pacientes afectados presenta una mutación en el gen SCN1A.
Los problemas comunes asociados con el síndrome de Dravet incluyen:
- Convulsiones prolongadas
- Convulsiones frecuentes
- Retraso en el desarrollo
- Afecciones ortopédicas
- Problemas odontológicos
- Problemas de lenguaje y habla
- Trastornos en el comportamiento y espectro autista
- Problemas de crecimiento y nutrición
- Dificultades para dormir
- Infecciones crónicas
- Trastornos de integración sensorial
- Dificultades en el movimiento y el equilibrio
- Deficiente funcionamiento del sistema nervioso autónomo
La edad de aparición de la enfermedad se sitúa entre los 4 y 12 meses de vida, caracterizándose por convulsiones clónicas o tónico-clónicas generalizadas o unilaterales de duración prolongada tanto en un contexto febril como en ocasiones en ausencia de fiebre.
En edades más avanzadas, es frecuente la aparición de otro tipo de crisis, como mioclonías, ausencias atípicas y parciales complejas. Otras comorbilidades como el retraso del desarrollo y los EEG anormales a menudo no son evidentes hasta el segundo o tercer año de vida.
Las opciones de tratamiento actuales son limitadas, y el cuidado constante requerido para una persona que padece el síndrome de Dravet afecta gravemente la calidad de vida del paciente y la familia.
Los pacientes con síndrome de Dravet enfrentan una tasa de mortalidad de alrededor del 15% debido a SUDEP (muerte súbita inesperada en la epilepsia), convulsiones prolongadas, accidentes relacionados con convulsiones como ahogamiento e infecciones.
Causas
Alrededor del 80% de los pacientes diagnosticados con síndrome de Dravet tienen una mutación en el gen SCN1A. Este gen codifica la producción de canales de iones de sodio, que son proteínas de poros incrustadas en la membrana celular que permiten que los iones de sodio entren y salgan de la célula, propagando señales eléctricas.
Algunos pacientes que parecen tener síndrome de Dravet tienen otras mutaciones, incluyendo SCN2A y PCDH19, que se corresponden con otros síndromes específicos.
El 90% de las mutaciones del SCN1A son de novo, lo que significa que no se ha heredado y no se encuentra en los padres del paciente.

Del 4 al 10% de las mutaciones del SCN1A se heredan de los padres. En este caso, hay un 50% de probabilidad de pasar la mutación a futuros niños.
Hay más de 6.000 posiciones para que ocurra una mutación en el gen SCN1A. Por lo tanto, la mayoría de las mutaciones de los pacientes no se han reportado en otras personas.
Se puede observar cualquier tipo de mutación SCN1A en el síndrome de Dravet, y el tipo de mutación no predice la gravedad de la enfermedad.
Las mutaciones en el gen SCN1A también se asocian con migrañas, convulsiones febriles (FS) o epilepsia generalizada con convulsiones febriles.
Cómo detectarlo
El síndrome de Dravet es un trastorno del neurodesarrollo que se caracteriza por una epilepsia severa resistente al tratamiento que presenta las siguientes características clínicas y electroencefalográficas:
- Inicio en el primer año de vida
- Desarrollo cognitivo normal previo al inicio de la crisis
- Resistencia al tratamiento farmacológico
- Crisis convulsivas prolongadas (más de diez minutos)
- Normalidad inicial con deterioro posterior del EEG, asociado a deterioro cognitivo progresivo con ataxia y otras alteraciones motoras
En la mayoría de los casos, las crisis epilépticas comienzan en el primer año de vida. Las primeras crisis están relacionadas con la aparición de fiebre y son convulsiones generalizadas tónicas-clónicas o hemiclónicas. En muchas ocasiones estas crisis desembocan en estatus epilepticus (episodios de larga duración o con repetición sin recuperación de la conciencia).
Con el tiempo, también aparecen crisis afebriles o relacionadas con otros estímulos, y otro tipo de convulsiones como las mioclonías, ausencias atípicas y crisis parciales-complejas.
A partir del segundo año se empiezan a observar síntomas de retraso en el desarrollo cognitivo y psicomotor. En muchos casos se observan ataxia, trastornos incluidos dentro del espectro autista, problemas alimenticios, de crecimiento y trastornos del sueño. El habla suele ser una de las facultades más afectadas.
Existe un alto porcentaje de casos que no cumplen todos los requisitos señalados. También se hallan otras formas de epilepsia de inicio clínico similar pero que no evolucionan de forma tan negativa.
Factores desencadenantes de las crisis en el síndrome de Dravet:
- Cuadro de fiebre
- Cambios bruscos de la temperatura corporal (baño, calor, ejercicio físico…)
- Determinados estímulos como patrones visuales, luces, etc.
- Emociones intensas
Fuente: Dravet Foundation





